2026-05-25
嵌合抗原受体(CAR)T细胞疗法在血液恶性肿瘤中疗效明确,但传统体外制备流程复杂、周期长、费用高昂(单次可达数十万美元),严重制约其普及。in vivo CAR-T工程作为变革性替代方案,可直接在患者体内重编程内源性T细胞生成CAR-T,无需细胞采集与体外扩增。来自《Cancer Research》的最新综述“Optimizing In Vivo CAR T-cell Engineering for Cancer Immunotherapy”系统梳理了当前in vivo CAR递送策略与临床转化进展,同时多项人体临床试验已取得突破性结果,标志着这一技术正从概念走向现实。
为何需要体内CAR-T?从“体外制备”到“体内生成”的范式革命
近十年来,所有获批CAR-T产品均采用自体T细胞体外制备流程:采血→体外激活→基因修饰→扩增→回输。尽管临床疗效确切,但制备周期长达数周、产品保质期短、需淋巴清除预处理、治疗费用极高,且高肿瘤负荷患者面临严重CRS与ICANS风险。
体内直接制备CAR-T为克服上述瓶颈提供了极具前景的方案。该策略无需体外细胞扩增与回输,以更平缓的动力学从少量转导T细胞群开始扩增,避免过度增殖导致的细胞耗竭。最新数据显示,这种低程度干预的CAR-T细胞可维持干细胞样表型,在远低于传统疗法剂量下即可发挥强效抗肿瘤效应。更重要的是,in vivo CAR-T直接利用患者内源性T细胞,免除淋巴清除化疗,降低感染并发症风险、维持免疫系统完整性,有助于促进表位扩散与更广泛的抗肿瘤应答,降低CAR靶点抗原逃逸风险。据估算,in vivo CAR递送单次剂量成本可低至5000美元。
当然,体内方案仍面临挑战:高肿瘤负荷患者存在免疫抑制风险,完整免疫系统可能对递送载体产生应答,脱靶递送会稀释治疗效果。该疗法成功取决于两大核心要素——高效T细胞靶向与高转导效率,二者是最大化疗效、最小化脱靶效应的关键。
目前,表面工程化慢病毒载体与脂质纳米粒(LNP)是最成熟、应用最广泛的递送系统。
病毒载体:精准靶向与多重工程改造
(一)常用病毒载体平台
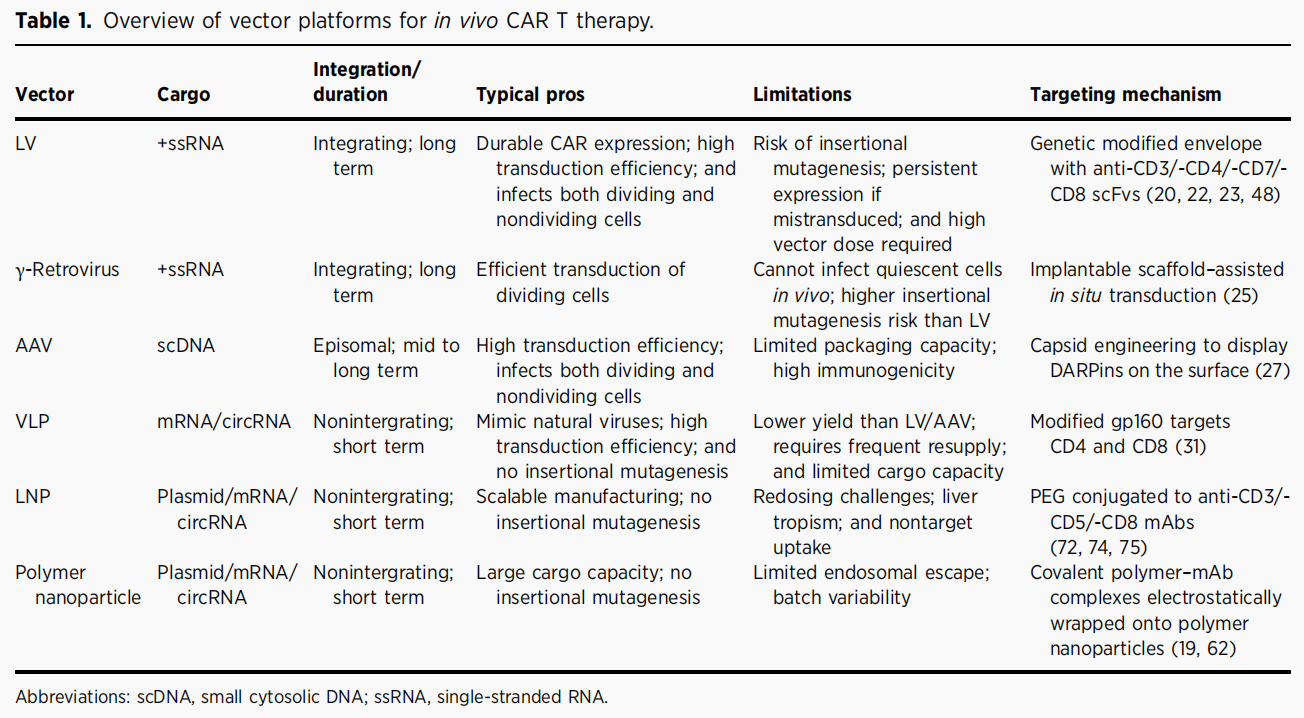
研究已使用多种复制缺陷型病毒载体实现in vivo CAR-T生成,包括慢病毒(LV)、γ-逆转录病毒、腺相关病毒(AAV)、病毒样颗粒(VLP),其中慢病毒应用最广泛。慢病毒可高效将大片段DNA整合至宿主基因组,可转导分裂期与非分裂期细胞,实现长效表达。
为将全身注射的慢病毒限定于循环T细胞,研究人员对载体进行了多重工程化改造。Pfeiffer等利用携带CD8+ scFv的尼帕病毒糖蛋白假型慢病毒,在人源化小鼠中实现T细胞选择性转导并有效清除CD19+细胞。Agarwal等开发CD4靶向慢病毒,在40-60%的CD4+淋巴细胞中实现CAR表达。Michaels等开发VivoVec平台,采用抗CD3 scFv重定向慢病毒,编码CD19 CAR与雷帕霉素诱导型合成细胞因子受体,实现T细胞靶向转导与可控扩增。
AAV以游离体形式维持长期表达,不整合宿主基因组。Theuerkauf等构建展示CD4-/CD32a结合DARPin的AAV2载体,优先转导双阳性细胞。Nawaz等采用靶向T细胞的AAV-DJ嵌合载体递送CD4 CAR质粒,在异种移植模型中清除人CD4+白血病T细胞。
VLP为非感染性颗粒,可包裹mRNA实现瞬时递送,大幅降低脱靶编辑与基因组整合风险。Wang等开发T细胞特异性融合VLP平台,利用突变gp160靶向CD4/CD8,在人源化淋巴瘤小鼠模型中实现瞬时CAR表达并控制肿瘤,且不转导非T细胞。
表1 In vivo CAR-T治疗的载体平台
(二)病毒表面修饰:重定向嗜向性
可通过在病毒包膜表面展示靶向分子重定向其嗜向性。主要策略包括:①基因工程将scFv整合至包膜蛋白(如VSV-G)的信号肽与跨膜结构域之间;②利用桥连抗体(蛋白A/G融合蛋白)或链霉亲和素-生物素系统进行非共价修饰;③化学点击反应将DBCO功能化抗体共价结合至含叠氮探针的病毒包膜。
与单克隆抗体相比,scFv操作简便、免疫原性低、生产成本低,且可整合共刺激分子实现体内CAR转导与T细胞活化同步完成,因此scFv假型病毒仍是主流方案。
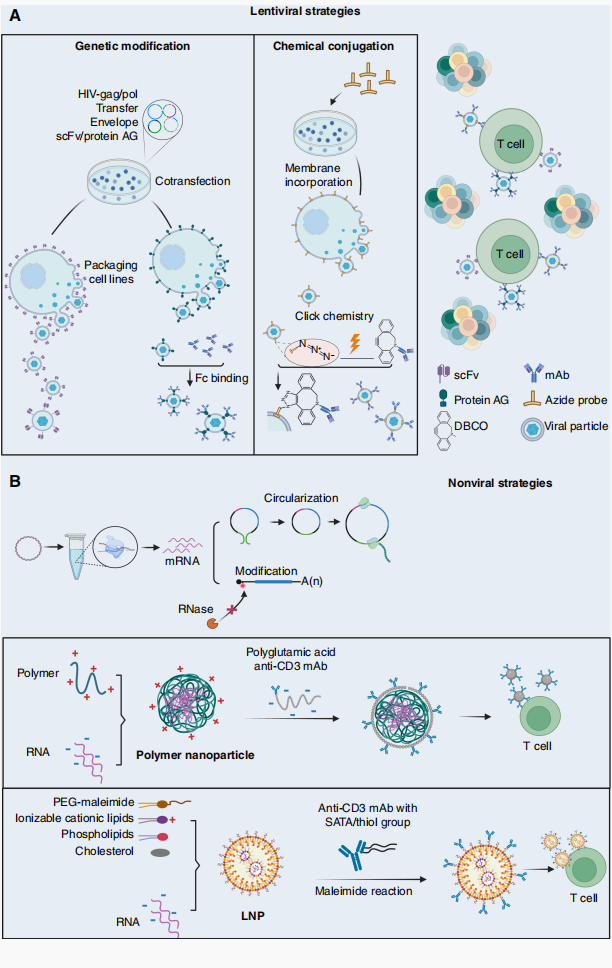
图1 通过慢病毒及非病毒递送系统实现靶向in vivo CAR‑T细胞工程的策略
(三)包膜蛋白选择:从VSV-G到新型副黏病毒
传统慢病毒采用VSV-G假型化,滴度高、稳定性强,但通过LDLR介导具有广谱嗜向性,且易被血清补体灭活,不适用于体内给药。
近期研究构建了对LDLR亲和力降低的VSV-G突变体(K47Q/R354Q、I182E),与scFv共表达时可实现T细胞选择性靶向。Cocal包膜糖蛋白与VSV-G同源性达71.5%,滴度相当但对人血清灭活抗性更强,基于该平台开发的体内CAR-T产品已进入临床研究。
副黏病毒糖蛋白(MV-H/F、NiV G/F)在细胞膜表面直接介导膜融合,无需内体逃逸。NiV假型慢病毒因抗NiV抗体血清阳性率低,不易被血清中和,已证实可在体内有效转导CD8+ T细胞并介导B细胞清除。Green等近期开发新型副黏病毒来源包膜蛋白,可高效转导小鼠"静息态"CD8+ T细胞并清除CD19+肿瘤。
非病毒载体:LNP引领的瞬时CAR表达新方向
(一)纳米颗粒系统
与病毒载体相比,纳米载体免疫原性更低、可规模化生产、支持重复给药。用于T细胞工程的阳离子聚合物(PBAE、PEI、PEG)通过静电相互作用与核酸自组装,表面正电荷提升细胞摄取,质子海绵效应促进内体逃逸。
脂质纳米粒(LNP)在体内T细胞重编程中展现巨大潜力。LNP通常由可电离阳离子脂质、辅助脂质、PEG化脂质组成。通过SATA-马来酰亚胺化学将靶向抗体修饰至LNP表面可提升T细胞特异性。Zhou等开发CD3抗体修饰LNP系统,在体内成功生成IL6敲低CAR-T细胞,在清除白血病的同时降低CRS。Carl June团队开发CD8靶向LNP搭载新型可电离脂质L829递送抗CD19 CAR mRNA,在啮齿类与灵长类模型中实现肿瘤控制与瞬时B细胞清除。
(二)mRNA vs 质粒DNA:为何mRNA成为主流
LNP介导CAR递送可采用mRNA或质粒DNA。DNA转基因表达时长更优、稳定性更高、成本更低,但需进入细胞核且存在非预期基因组整合风险。mRNA仅需到达细胞质即可直接翻译,无基因组整合风险,且可通过体外转录快速规模化生产,因此已成为LNP介导CAR递送的主流选择。
为克服mRNA固有免疫原性,研究采用掺入假尿苷等修饰核苷、优化5’帽结构与3’ poly(A)尾等策略显著提升蛋白表达。环状RNA(circRNA)作为新一代替代方案,共价闭合结构稳定性更高、免疫原性更低,可携带多个开放阅读框,已有临床试验采用circRNA平台进行体内CAR递送。
超越T细胞:NK细胞与巨噬细胞的体内CAR工程化
CAR-T主要用于血液肿瘤,而CAR-巨噬细胞(CAR-M)已成为实体瘤治疗的潜力方案。CAR-M可浸润肿瘤微环境,将M2型重编程为M1型,降低TAM比例并增强抗原呈递。Liu等采用甘露糖修饰LNP向胰腺癌瘤内巨噬细胞递送MUC1-CAR DNA,31%巨噬细胞被重编程,显著延长小鼠生存期。
Yang等采用可电离脂质LNP向肝癌肝巨噬细胞共递送GPC3 CAR与Siglec-GΔITIMs的mRNA,24小时内肝脏30%巨噬细胞呈双阳性,引发强烈免疫应答。
CAR-NK研究尚处起步阶段。多数报道的CAR-NK为体内CAR-T工程中的伴随产物。Agarwal等递送NiV假型化CD8-慢病毒,在骨髓与脾脏意外产生CAR-NK与NKT细胞,虽仅占总群体1-2%,但在肿瘤部位富集。LNP-mRNA是CAR-NK的替代策略,无需NK细胞激活且无基因组整合风险,但体内递送仍处探索阶段。
靶向分子决定转导效率
体内CAR递送中,scFv或mAb的选择不仅决定靶细胞类型,还影响转导效率与安全性。核心原则包括:①靶抗原需高丰度、特异性表达,如CD8靶向慢病毒也会产生少量CAR-NK,但溢出效应可能有益;②需考虑靶分子内吞能力,Chen等研究显示CD4/CD5靶向LNP虽结合率仅低10%–50%,但eGFP表达降低约90%;③需平衡激活潜能与CRS风险,抗CD3 scFv可激活静息T细胞但过度激活可能导致耗竭,CD5负调控TCR信号因此CRS风险最低。
体内递送效率是内吞驱动增益与激活相关风险的平衡结果。Billingsley等研究显示CD3-LNP阳性率(15-17%)高于CD7-LNP(5-6%);Coradin等发现CD3-慢病毒体内效率(峰值55%)高于CD8-慢病毒(35%),可能与CD3刺激诱导扩增相关。
临床研究进展:从概念验证到人体突破
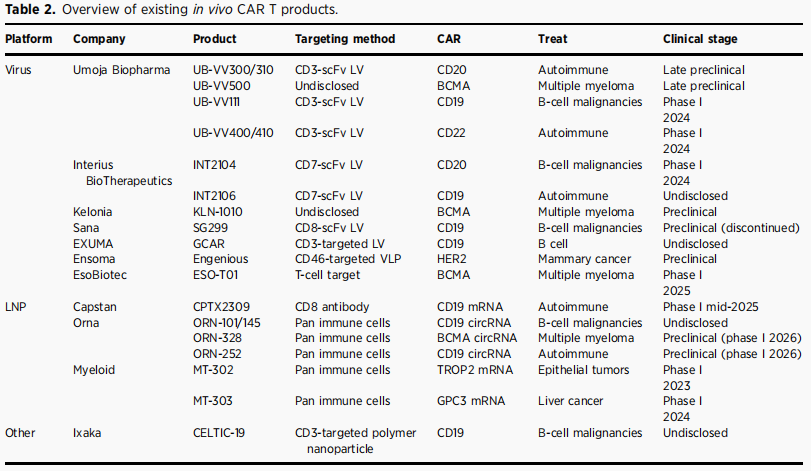
目前体内CAR-T临床产品分为两类:慢病毒载体(永久改造)代表企业包括Umoja、Interius、EsoBiotec;mRNA-LNP(瞬时表达)代表企业包括Capstan、Myeloid、Orna。
Interius BioTherapeutics的INT2104于2024年7月成为全球首个进入人体临床试验的体内CAR疗法(Ⅰ期),为靶向CD7+ T细胞与NK细胞的抗CD20 CAR慢病毒,16只食蟹猴中15只循环B细胞降低≥75%。
Umoja Biopharma的VivoVec平台采用展示多结构域融合蛋白(CD58+抗CD3 scFv+CD80)的第三代慢病毒,增强T细胞活化与共刺激,UB-VV111已获FDA IND批准进入Ⅰ期临床。
EsoBiotec的ESO-T01为首个进入临床的BCMA靶向体内CAR-T,2025年1月在中国启动Ⅰ期试验。2025年7月《柳叶刀》简报报道:最低剂量组前4例患者中2例严格完全缓解、2例部分缓解,所有4例第28天MRD阴性。这项发表于《Nature Medicine》的研究共入组5例复发难治多发性骨髓瘤患者,单次静脉输注0.2×109TU,全程无需清淋、无需采血、无需体外制备,入组至输注中位时间仅8小时。ORR达80%(4/5),其中3例sCR、1例VGPR,所有缓解患者第60天MRD阴性,中位起效时间仅15天。安全性整体可控,CRS发生率80%(均为1-3级,经糖皮质激素或托珠单抗快速缓解),ICANS仅1例1级。研究首次揭示双相免疫激活特征:0-24小时为先天免疫波(IL-6、TNF等升高),第6-14天为适应性免疫波(IFNγ、IL-10等升高),与CAR-T峰值扩增同步,为临床毒性管理提供了重要依据。
Capstan的CPTX2025年6月在澳大利亚完成首例健康志愿者给药,其CPTX2309采用CD8靶向LNP递送抗CD19 CAR mRNA,体外研究显示高达80%的CD8+ T细胞表达CAR,B细胞深度清除提示免疫重置效应。
Create Medicines的MT-302为首个进入临床的体内CAR疗法,采用CD89信号驱动的TROP2靶向CAR mRNA-LNP,功能性CAR表达仅限于FcRγ+髓系细胞,已在乳腺癌模型中展现抗肿瘤活性。MT-303编码GPC3靶向CAR用于肝癌,首例患者已于2024年8月接受治疗。
Wang等报道的HN2301(CD8靶向LNP包裹CD19 CAR mRNA)治疗难治性系统性红斑狼疮早期结果显示:给药后6小时内约5-50%的循环CD8+ T细胞表达CAR,低剂量组2例B细胞部分清除、高剂量组3例完全清除,未观察到3级或4级CRS与ICANS。
表2 现有in vivo CAR‑T产品概览
总结与展望
尽管进展迅速,in vivo CAR-T仍面临多重挑战:①淋巴清除仍是关键瓶颈,但近期非人灵长类研究证实单次CD3靶向慢病毒无需淋巴清除即可诱导in vivo CAR-T扩增并维持B细胞清除长达10周;②慢病毒存在基因组整合风险,需通过T细胞特异性启动子、miRNA靶位点等策略限制脱靶表达;③LNP基因转移效率低于病毒系统,且重复给药可能触发抗LNP抗体产生;④LNP无法生成记忆表型CAR-T细胞,能否实现持久肿瘤控制仍需验证。
脱靶转导是共同挑战,慢病毒自身可展示CAR货物转导CD19+ B细胞,需开展专项监测。缓解策略包括采用支架局部递送——将装载慢病毒或mRNA-LNP的可注射水凝胶植入肿瘤附近,在病灶部位实现空间靶向、控释、降低全身暴露,已在多个临床前模型中证实可行。
展望未来,in vivo CAR技术最具变革性的价值在于可重编程传统体外工程难以触及的免疫细胞——短寿命中性粒细胞、固有淋巴细胞、特异性树突状细胞等,有望解锁免疫治疗全新维度。随着多款产品进入临床试验,建立完善的监管框架与伦理指南至关重要,最终推动in vivo CAR-T实现更广泛的临床应用,让“一次输液、体内造CAR-T”从梦想走向现实。

地址:中国武汉东湖高新区光谷七路128号 市场:17720522078 人事行政:027-62439686 邮箱:marketing@genevoyager.com
BD 商务总台:13886000399(BD 经理)
本公司所有产品仅供实验科研使用,不用于人体疾病治疗及临床诊断。
地址:中国武汉东湖高新区光谷七路128号 市场:17720522078 人事行政:027-62439686 邮箱:marketing@genevoyager.com
BD 商务总台:13886000399(BD 经理)
本公司所有产品仅供实验科研使用,不用于人体疾病治疗及临床诊断。
